Micro-dystrophin: A Small Gene With Big Promise
Micro-dystrophin: A Small Gene With Big Promise https://pediatricsnationwide.org/wp-content/uploads/2023/04/Spring-23-Cover-Final-crop-1024x632.jpg 1024 632 Abbie Miller https://pediatricsnationwide.org/wp-content/uploads/2023/05/051023BT016-Abbie-Crop.jpg
SRP-9001 for Duchenne muscular dystrophy supplies a functional dystrophin gene via AAVrh74 gene therapy.
In 1969, Jerry Mendell, MD, was working at the National Institute of Neurological Disorders and Stroke (NINDS) when he saw his first patient with Duchenne muscular dystrophy (DMD).
DMD, a severe form of muscular dystrophy caused by a mutation in the DMD gene and resulting in a lack of the dystrophin protein, leads to muscle weakness and atrophy. DMD is an X-linked condition and, as such, typically affects boys.
Since that day, Dr. Mendell has been on a mission to develop new treatments for patients with DMD and other neuromuscular diseases. His track record is impressive, with advances in understanding how corticosteroids can improve outcomes for children with DMD and pioneering work in adeno-associated virus (AAV)-mediated gene therapy. He led the team behind the first systemically delivered AAV gene therapy to be approved by the U.S. Food and Drug Administration (FDA) — Zolgensma®, which offers life-changing outcomes for children with spinal muscular atrophy (SMA).
His next achievement may bring him full circle — back to offering a potentially life-changing therapy for children with DMD.

“Since I began investigating gene therapy as a potential treatment for children with neuromuscular disorders, it’s been my dream to develop a gene therapy for DMD,” says Dr. Mendell, neurologist and principal investigator in the Center for Gene Therapy in the Abigail Wexner Research Institute at Nationwide Children’s Hospital. “To this point, we’ve had very little to offer these families. Until recently, our only approved treatment for Duchenne muscular dystrophy has been glucocorticoids, such as prednisone. Our research aims to see if gene therapy is a safe and effective option for these patients and others with rare neuromuscular diseases in the future.”

Delandistrogene moxeparvovec, known as SRP-9001, which Dr. Mendell co-invented with Louise Rodino-Klapac, PhD, formerly at Nationwide Children’s and presently the executive vice president, head of R&D and chief scientific officer at Sarepta Therapeutics, is a novel DMD gene-replacement therapy currently in clinical trials. Sarepta Therapeutics licensed this gene therapy program from Nationwide Children’s in 2018, and recently filed a Biologics License Application (BLA) with the FDA in the fall of 2022.
Building the Therapy That Would Become SRP-9001
Gene therapy for DMD has been a long time coming. So, Dr. Mendell always begins his talks with a history lesson.
Gene therapy was first proposed by Theodore Friedman about 50 years ago, with the first clinical trials beginning in the 1990s. Gene therapy research nearly ground to a halt in 1999 after the tragic and high-profile death of a patient named Jesse Gelsinger and a rash of leukemia cases linked to insertional mutations caused by retrovirus vectors. Gelsinger was treated with an adenoviral gene therapy and died because of a massive inflammatory response. In light of these setbacks, the future of gene therapy looked bleak.
Hope for gene therapy was renewed when researchers began using adeno-associated virus (AAV), instead of adenovirus or retrovirus, vectors as the vehicle of choice for systemic gene therapy. AVV is a small virus, about 25 nm, made of a nonenveloped protein shell and containing a linear single-stranded DNA genome of about 4.7 kb (kilobase, a measure of length equal to 1,000 base pairs).
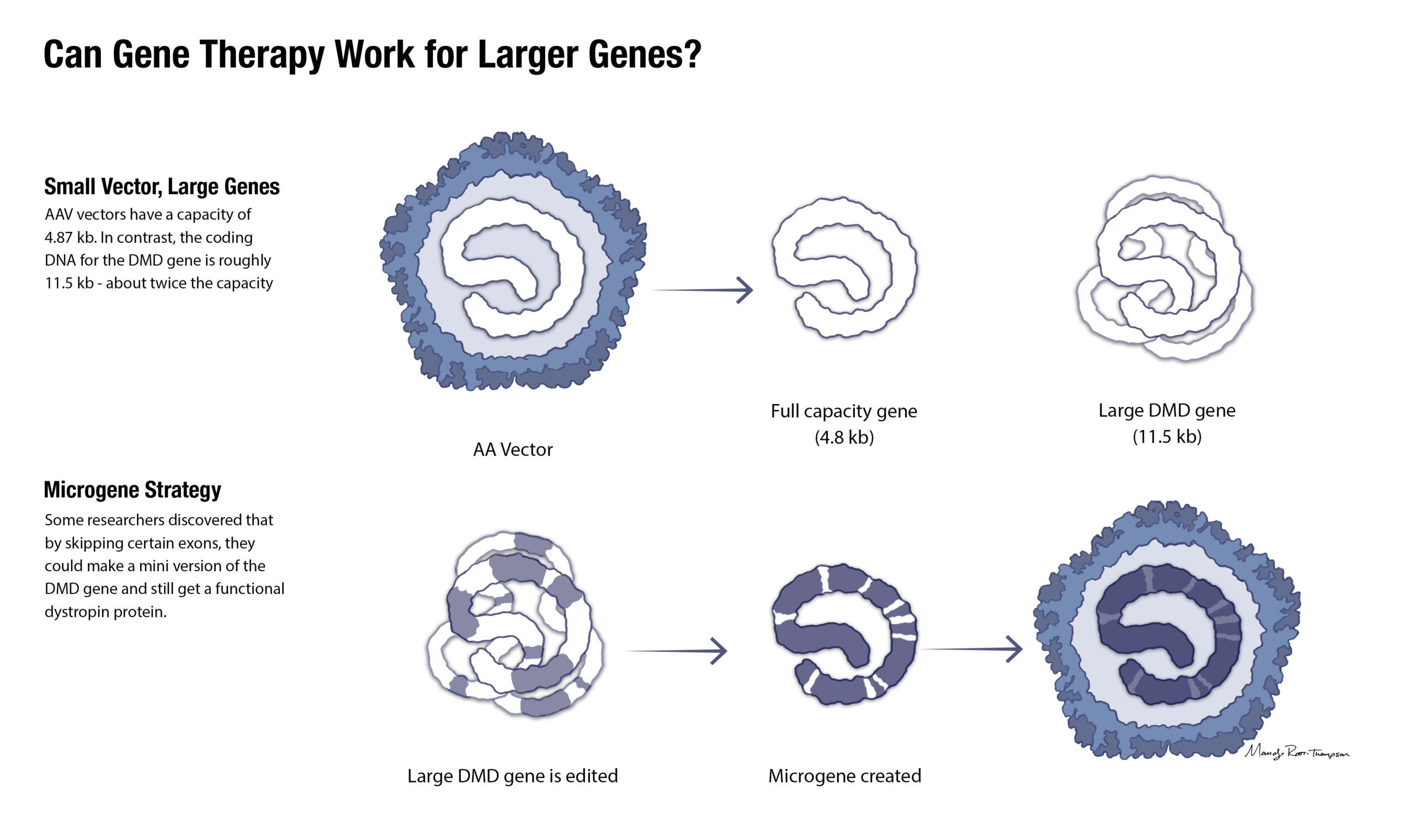
One of the challenges of developing an AAV-mediated gene therapy for DMD is the sheer size of the DMD gene. The DMD gene’s cDNA is about 11.5 kb. That’s more than twice as large as the vector’s capacity. With the required promoter that activates the gene, the transgene is 14 kb.
Drs. Mendell and Rodino-Klapac developed a shortened functional dystrophin gene, a micro-dystrophin, that would fit inside the AAV vector and produce a dystrophin protein that contains functional components of wild-type dystrophin in order to provide clinical benefit.
Their proposed therapy would work like this: when delivered via AAV vector, the micro-dystrophin gene would enter the muscle cells and be translated into a functional protein, which was missing from the DMD muscle cells.
To determine if the idea would work, preclinical studies were done to show if administering the therapy intravenously would change the amount of dystrophin in the muscle cells. If they administered the therapy and the muscle cells did not show increased dystrophin — they would be back to the drawing board.
Dr. Rodino-Klapac has been working on this therapy for nearly two decades. She says one of the first moments that stands out in her memory of working on the therapy was the first time she observed SRP-9001 protein expression localized to the membrane of muscle fibers.
“I recall running to bring Dr. Mendell to the microscope room to share in the moment,” she says.
The therapy they developed uses AAVrh74 as the vector, MHCK7 as the promoter, and a shortened functional SRP-9001-dystrophin gene. The shortened functional gene was inspired by a 2002 Nature publication by Scott Harper, PhD, and team reporting on internal truncated dystrophin genes found in Becker muscular dystrophy patients. Dr. Harper’s work eventually led him to other areas of science — specifically studying RNA interference as a therapy for dominantly inherited genetic disorders — but his early research on various small dystrophins has persisted.
“It’s a fun fact that I like to share,” says Dr. Mendell. “Dr. Harper works at Nationwide Children’s now, and though we never collaborated on this gene therapy product, and our research focuses on very different diseases and approaches, there’s this connection.”
“We used AAVrh74 for a few reasons,” says Dr. Mendell. “First, we had isolated it here at Nationwide Children’s, so it was accessible. Second, it had very good distribution into all types of muscle, including skeletal, heart and diaphragm. Third, as an AAV derived from non-human primates, the percentage of the population with pre-existing antibodies is low.”
In an early safety study of a mini-dystrophin gene therapy at Nationwide Children’s, the team discovered an unexpected wrinkle that could thwart their hopes for gene therapy success.
The discovery was published in 2010 in the New England Journal of Medicine. A patient participating in the gene therapy trial had a large DMD gene mutation with deletion of exons 3-17. The mini-gene used in this trial was delivered to the patient’s arm muscle. When the gene expressed the mini-dystrophin protein, an immune response was elicited. An important lesson for future trials: do not transfer a gene that can express a novel protein and cause an immune response from the patient. For investigational DMD gene therapies, patients with exon 3-17 deletions were frequently excluded from clinical trials.
Bringing SRP-9001 to Clinical Trial
SRP-9001-101 was the first phase 1 study of a systemic gene therapy for DMD. It began in 2018, and the first year results were published in JAMA Neurology in 2020. The rationale for the study used evidence from preclinical studies as well as learnings from the then newly approved Zolgensma.
Dosing was set at 2×1014 vg/kg (vector genomes per kilogram of bodyweight) for the first trial. This is the same dosing as used in Zolgensma, but Zolgensma is only approved in infants and children up to 2 years of age. Using this dosing for older children required a lot more vector and potentially increased the risk of toxicity.
“This is a huge amount of vector,” says Dr. Mendell. “Groups that are still using AAV9 are experiencing trouble with toxicity at this level. Our choice to use AAVrh74 has turned out to be a big piece of our ability to minimize toxicity.”
The first trial targeted children aged 4 to 7 years. This is the age range when DMD symptoms really start showing. While the evidence points to “the earlier the better” for gene therapy for SMA, treating infants with DMD would have required nearly a decade of observation to find out if it worked. By treating children who were not yet or just beginning to show symptoms and following them for five years, the research team would have the fastest, safest and most efficient way to test the effect of the therapy on clinical outcomes.
Finally, because of a potential risk for transgene immunity, participants with mutations exclusively in exons 18-58 were included.
“Our first trial showed robust micro-dystrophin in the muscle fibers of the calf muscle in all participants,” says Dr. Mendell. “And even more exciting, they experienced no toxicity!”
“When we saw the first signs of clinical functional benefit in patients with Duchenne, it was a truly rewarding, humbling experience,” adds Dr. Rodino-Klapac.
After the initial success of the 101 clinical trial, the researchers were excited to expand their work.
Expanding Clinical Studies and Gathering Results
Additional studies of SRP-9001, including SRP-9001-102, SRP-9001-103 (ENDEAVOR), and SRP-9001-301 (EMBARK), are underway. EMBARK is a global, randomized double-blind placebo-controlled study with results expected at the end of 2023. An additional study in older and non-ambulant individuals with Duchenne, Study SRP-9001-303 (ENVISION), is planned to start later in 2023.
“Through our clinical development program for SRP-9001, Sarepta has dosed over 140 individuals with Duchenne across a wide range of disease progression,” says Dr. Rodino-Klapac. “As a result, we have a comprehensive data package with biologic and clinical data from multiple studies of SRP-9001. The data shows a compelling story of the safety, efficacy and durability of SRP-9001.”
Across all the clinical studies to date, the safety profile of SRP-9001 has remained consistent and manageable, she adds.
After validating the expression of the micro-dystrophin in the muscle tissue, the researchers have also monitored functional improvements.
One tool that is the established best practice for measuring motor function is the North Star Ambulatory Assessment (NSAA). The 17-item rating scale evaluates how well a child can perform a range of functions, including walking, running and getting up from a reclined or
lying position.
Across three independent SRP-9001 studies, the team saw meaningful and statistically significant improvements in NSAA, ranging from a 3.8-point improvement at 1-year post-dose to a 9.9-point improvement, when compared to the propensity-score-weighted external control, at 4 years post-dose, according to Dr. Rodino-Klapac.
The 3.8-point improvement was observed in study SRP-9001-103, an open-label clinical trial, and among a cohort comprised of patients ages 4 to 7 who are ambulatory. The nearly 10-point improvement was observed in study SRP-9001-101, the initial single-center
study.

“This data is especially meaningful to me because Duchenne is a disease that gets progressively worse,” says Dr. Rodino-Klapac. “Around age 9, these boys would typically be in the steep decline phase of their disease, losing skills and declining about three NSAA points per year. But instead of declining, the data showed the boys in that study not only increased their function but maintained the higher score over the course of the 4-year observation.”
“I have been with all of these trials from the start. It has been very exciting to see the progression as we have moved through the studies. We started with boys ages 4-7 with select mutations. Since then, we have been able to include in clinical trials younger and older boys with a wider variety of mutations, as well as ambulatory and non-ambulatory boys. We have worked with so many wonderful families and it has been a truly gratifying experience,” says Kelly Lehman, MSN, CRN, a research nurse practitioner in the Center for Gene Therapy.

Next Steps
Study SRP-9001-303 will have boys older than 8 years old who are both ambulatory and non-ambulatory.
“While most of the SRP-9001 trials have focused on younger boys, we are exploring the safety and
effectiveness of the therapy in patients who are older or non-ambulatory,” says Dr. Rodino-Klapac. “Our aim is to expand the availability of SRP-9001 to a larger percentage of the Duchenne population.”
To even further expand the number of patients who could potentially benefit from the therapy, finding a way to narrow the early-exon mutation exclusions is a logical next step. According to Dr. Rodino-Klapac, Sarepta has already begun a study to do just that.
“We are investigating what our research informs us are low-risk mutations in the currently excluded range,” she says.
Additional work is focused on expanding access to the therapy for people who have pre-existing antibodies to the viral vector used in the gene therapy.
“SRP-9001 uses AAV to deliver missing or corrected genes to cells. Patients who have pre-existing IgG antibodies are not currently eligible for treatment because the antibodies interfere with the therapy’s ability to reach the target cells and work as designed,” Dr. Rodino-Klapac says.
According to Dr. Rodino-Klapac, approximately 14% of patients with DMD have pre-existing AAV antibodies directed against the AAV used in SRP-9001. To meet this population’s needs, her team is studying a potential pre-treatment that could inhibit the reaction of those IgG antibodies.
If this pretreatment is successful, it could offer a way to expand the accessibility of other AAV-based gene therapies to patients for whom the promise of gene therapy remains out of reach.
Waiting for the FDA
The FDA decision, which could mark the first approved gene therapy for the treatment of DMD, is expected in late May. While families await news of approval and what restrictions will be set around the therapy’s use, Lehman says that health care providers can offer important insight and communication with families interested in participating in trials or getting a (hopefully) commercially available therapy in the future.
“These families so desperately want to find significant treatments for their boys with Duchenne muscular dystrophy and give them a longer life with better quality of life,” says Lehman. “SRP-9001 appears to be significant, based on study results published so far. Providers can help to manage expectations of what the potential is for the individual child and continue to manage their overall care needs.”
Dr. Mendell is looking forward to a brighter future for children and families with DMD.
“At the end of the day, where we are in this field is something I’m incredibly proud of. We’ve accomplished a lot in gene therapy in the last few years, and while there’s still work to do — expanding newborn screening for DMD, addressing treatment options for patients with preexisting antibodies and eliminating the exon exclusions — we’re on our way,” says Dr. Mendell. “I think the future looks bright. We’re very excited to see what happens next.”
Infusion Day
Infusion protocols for gene therapies have evolved as the trials have proceeded. While the first SRP-9001 infusions at Nationwide Children’s were done in the pediatric intensive care unit, they are now performed as an outpatient procedure. The infusion process typically takes under two hours. Continuous monitoring and supportive care helps ensure safe and successful delivery.
At Nationwide Children’s, the clinical research team has administered more doses of systemic gene therapy than perhaps anywhere else. As a result, they have a system that has been refined by experience and expertise.
When a child comes in for gene therapy at Nationwide Children’s, they have an IV placed in each hand. One is used for saline to maintain hydration, since participants are typically told not to eat or drink for four hours before the infusion.
The other IV is for the infusion. The added bonus of the second IV is that if one fails, the infusion can be moved over to the second without disrupting the infusion time.
The infusion at Nationwide Children’s typically takes 60-90 minutes. Slower infusions typically produce more optimal results, according to Dr. Mendell.
The team at Nationwide Children’s also has a protocol for their patients that includes exercising the extremities during the infusion. By gently moving the shoulders, elbows, hips, knees and ankles every 10 minutes in rotation during the infusion, the team enhances by increasing the blood flow to the exercising muscles, says Dr. Mendell.
“Our center’s results have been excellent, and we hope to be able to share our experience so that our protocols and techniques can help children receiving their infusions elsewhere,” says Dr. Mendell.
This feature was published in the Spring/Summer 2023 issue. Download the full issue.
References:
- Asher DA, Thapa K, Dharia SD, Khan N, Potter RA, Rodino-Klapac LR, Mendell JR. Clinical development on the frontier: gene therapy for Duchenne muscular dystrophy. Expert Opinions on Biological Therapeutics. 2020 Mar;20(3):263-274.
- Goedeker NL, Dharia SD, Griffin DA, Coy J, Truesdale T, Parikh R, Whitehouse K, Santra S, Asher DR, Zaidman CM. Evaluation of rAVVrh74 gene therapy vector seroprevalence by measurement of total binding antibodies in patients with Duchenne muscular dystrophy. Therapeutic Advances in Neurological Disorders. 2023 Jan 24;16:17562864221149781.
- Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, Chamberlain JS. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nature Medicine. 2002 Mar;8(3):253-261.
- Mendell JR, Sahenk Z, Lehman K, Nease C, Lowes LP, Miller NF, Iammarino MA, Alfano LN, Nicholl A, Al-Zaidy S, Lewis S, Church K, Shell R, Cripe LH, Potter RA, Griffin DA, Pozsgai E, Dugar A, Hogan M, Rodino-Klapac LR. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurology. 2020 Sep 1;77(9):1122-1131.
Image credits:
- Jerry Mendell, Nationwide Children’s
- Illustrations by Mandy Root-Thompson, for Nationwide Children’s
- Louise Rodino-Klapac, courtesy of Sarepta
- Kelly Lehman, courtesy of Kelly Lehman
About the author

Abbie (Roth) Miller, MS, MWC, is a passionate communicator of science. As the manager of medical and science content at Nationwide Children’s Hospital, she shares stories about innovative research and discovery with audiences ranging from parents to preeminent researchers and leaders. She is a Medical Writer Certified®, credentialed by the American Medical Writers Association, and received her masters of science in Health Communication from Boston University.
- Abbie Millerhttps://pediatricsnationwide.org/author/abbie-miller/

- Abbie Millerhttps://pediatricsnationwide.org/author/abbie-miller/
- Abbie Millerhttps://pediatricsnationwide.org/author/abbie-miller/
- Abbie Millerhttps://pediatricsnationwide.org/author/abbie-miller/