Treating Sickle Cell Disease in the United States
Treating Sickle Cell Disease in the United States https://pediatricsnationwide.org/wp-content/uploads/2021/03/AdobeStock_137900548-sickle-cell-header-1024x575.gif 1024 575 Abbie Miller https://pediatricsnationwide.org/wp-content/uploads/2023/05/051023BT016-Abbie-Crop.jpg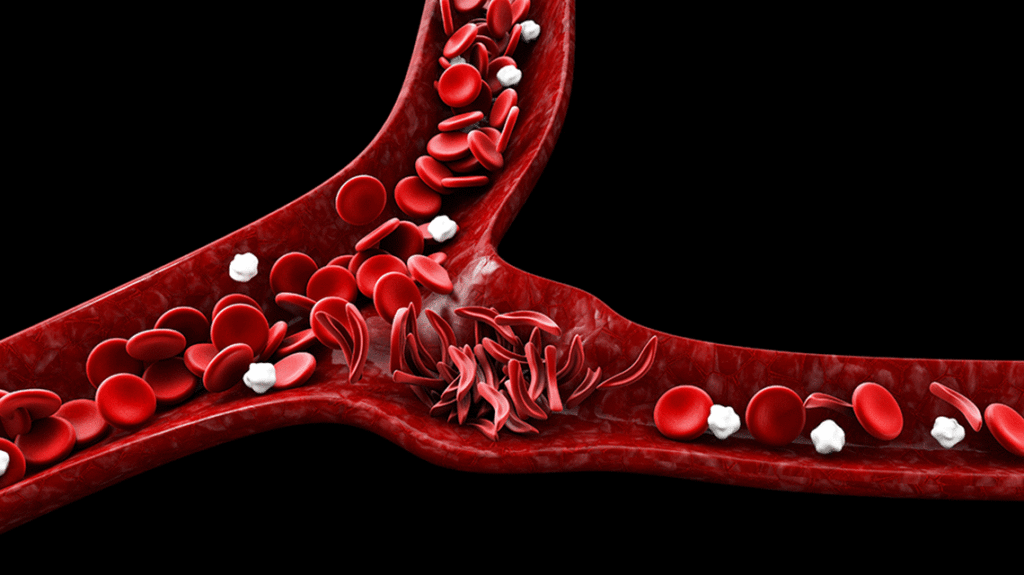
While people with sickle cell disease have better outcomes in the United States and other Western countries, progress in treating the disease has been slow moving.
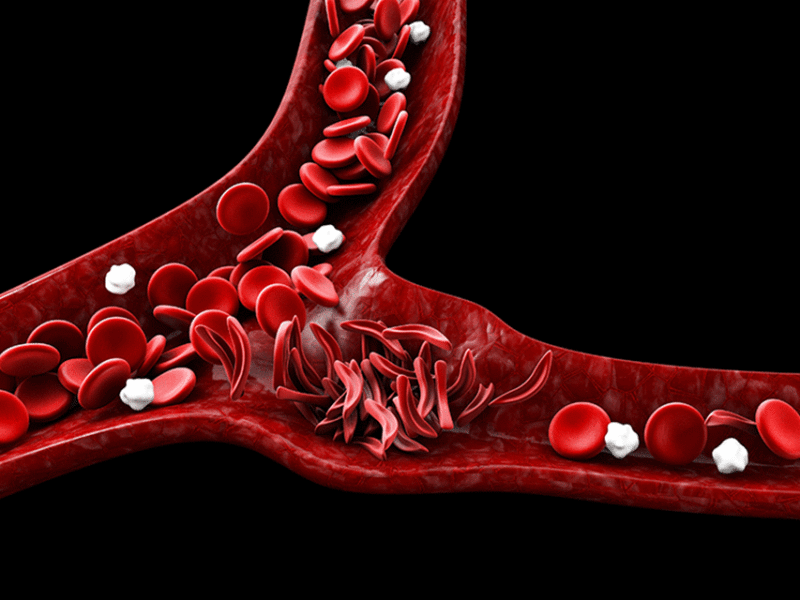
Sickle cell disease is marked by painful sickle cell crises, in which sickle-shaped cells get distorted in the small vessels and cause problems including disrupting blood flow and a lack of oxygen at the tissue level — microinfarcts — leading to severe pain.
Limited drug choices, gaps in education and a need for comprehensive care comprise serious challenges for patients and practitioners dealing with sickle cell disease. In an age in which research funding is highly competitive, it can also be a challenge to get support for sickle cell disease research and drug development.

Treating Sickle Cell Disease With Drugs
Only one drug — hydroxyurea — is FDA-approved to help reduce the number of crises a person with sickle cell disease experiences. When taken at clinical-trial level doses, it has been shown to be very effective. But there are barriers to using hydroxyurea in children with sickle cell disease, and a recent study published in BMC Research Notes finds that caregivers frequently decline the drug for their children.
“Even though all caregivers surveyed in the study noted that sickle cell disease negatively impacted daily life for the caregivers and the patients, not all caregivers choose to give hydroxyurea to their children,” says Susan Creary, MD, physician in the Division of Hematology/Oncology/BMT at Nationwide Children’s Hospital and lead author on the study.
According to the study, reasons for choosing not to give hydroxyurea included belief that their children did not have “severe” sickle cell disease. Caregivers who did not choose hydroxyurea also did not ask their child’s provider questions about the medication when they had fears about it.
Additionally, Dr. Creary explains that better efforts to educate caregivers about sickle cell disease and hydroxyurea are needed to enable caregivers to make informed choices. “Unfortunately, caregivers are not always educated by the health care team,” Dr. Creary says. “When they get their information from the community and the internet a lot of misinformation can play into their decision making.”
Beyond educating patients and families about hydroxyurea and sickle cell disease is the need for more treatment options.
Even when patients are taking hydroxyurea, they may still have pain crises. And once a patient is in crisis, the only available treatment is pain management. But this could change in the near future.
A new drug currently awaiting phase III clinical trial could change that. In the phase II randomized, double-blinded study of the efficacy, safety and pharmacokinetics of Rivipansel, patients who received the drug experienced shorter length of hospital stay, shorter time to crisis resolution and reduced use of opioids for pain management.
This drug has been a long time coming.
Since the signing of the Sickle Cell Anemia Control Act in 1972, the prognosis for children born with sickle cell disease has improved, but much of that has come from better supportive care, more widely available vaccines and the use of prophylactic antibiotics, explains Anthony Villella, MD, physician in the Division of Hematology/Oncology/BMT at Nationwide Children’s.
There is still a lot of work to be done. Historically, drug companies and research institutions have neglected sickle cell disease and instead focused on diseases treated with lucrative medications.

“For example, with hemophilia, another rare hematological disease, you have an abundance of therapeutic options because for many years there has been fierce competition among drug companies to bring very effective, but also very expensive, drugs to market. Consequently, most patients with hemophilia now can enjoy a near normal quality of life,” says Dr. Villella. “Our hope is that someday this will be true for sickle cell disease, as well.”
Life-Long, Comprehensive Care
As the patient ages, the length of each sickle cell crises tends to increase. The average hospital stay is four to five days, but most, an estimated 90 percent, of crises are treated at home with over-the-counter and prescription medications. “And just because a patient is discharged doesn’t mean the crisis is over,” explains Dr. Villella. “Most patients are sent home with medications to help them continue to manage the pain.”
Patients with sickle cell disease often rely on Emergency Departments for care during a crisis. Because opioid medications are frequently required to treat pain crises, adolescent and adult patients are often stigmatized and regarded as drug seekers, even when they reveal their sickle cell diagnosis.
While doctors and patients wait for more drug options for treating sickle cell disease, the focus is on providing comprehensive support and education for patients, families and communities.
The psychological complications associated with sickle cell disease are largely the result of having a genetic disease that causes severe, chronic pain and other symptoms that interfere with the daily life of the patient. The problems most frequently encountered, according to a review in the British Journal of Haematology, are anxiety, depression, social withdrawal, aggression, poor relationships and poor school performance.
Nationwide Children’s Sickle Cell Comprehensive Clinic serves children, adolescents and adults who have sickle cell disease. During each clinic visit, the patients are seen by a physician, nurse practitioner, nurse clinician, social worker, psychologist, dentist and genetic counselor. This integrated approach is vital to creating a support system for the patients and their families.
“Sickle cell disease affects a disproportionate number of people from minority ethnic groups, many of whom also struggle with poverty and lack of insurance and access to care. Hospitals are not going to make money off of this patient population. We are fortunate to have an extraordinarily dedicated team and the support of Nationwide Children’s Hospital for the care we provide,” says Dr. Villella.
References:
- Creary S, Zickmund S, Ross D, Krishnamurti L, Bogen DL. Hydroxyurea therapy for children with sickle cell disease: Describing how caregivers make this decision. BMC Research Notes. 2015 Aug 25;8:372.
- Anie KA. Psychological complications in sickle cell disease. British Journal of Haematology. 2005 Jun;129(6):723-729.
Photo credit: Nationwide Children’s
About the author

Abbie (Roth) Miller, MS, MWC, is a passionate communicator of science. As the manager of medical and science content at Nationwide Children’s Hospital, she shares stories about innovative research and discovery with audiences ranging from parents to preeminent researchers and leaders. She is a Medical Writer Certified®, credentialed by the American Medical Writers Association, and received her masters of science in Health Communication from Boston University.
- Abbie Millerhttps://pediatricsnationwide.org/author/abbie-miller/

- Abbie Millerhttps://pediatricsnationwide.org/author/abbie-miller/
- Abbie Millerhttps://pediatricsnationwide.org/author/abbie-miller/
- Abbie Millerhttps://pediatricsnationwide.org/author/abbie-miller/