When Every Week Matters: Advancing a Treatment to the Clinic
When Every Week Matters: Advancing a Treatment to the Clinic https://pediatricsnationwide.org/wp-content/uploads/2021/10/Learning-in-Real-Time-e1633354760908-1024x491.jpg 1024 491 Natalie Wilson https://pediatricsnationwide.org/wp-content/uploads/2021/06/Natalieheadshot3-2.png
Researchers and regulatory experts bring a potential new therapy for a deadly neurodegenerative disease from IND application to clinical trial enrollment in a matter of weeks, setting a new standard in translating therapies from bench to bedside.
In October 2021, the Office of Research Regulatory Affairs (ORRA) at Nationwide Children’s Hospital submitted an Investigational New Drug (IND) application to the U.S. Food and Drug Administration (FDA) for gene therapy AAV9.IGHMBP2.
Within the same month, the ORRA received official notice that a clinical trial could begin. The first patient was enrolled by November.
“The ability to begin enrolling patients so quickly is really a huge testament to the strengths of Nationwide Children’s,” says Kathrin Meyer, PhD, a principal investigator in the Center for Gene Therapy in the Abigail Wexner Research Institute (AWRI) at Nationwide Children’s who led the preclinical work to develop the AAV9 gene therapy. “All the committees that needed to sign off after the FDA cleared the IND were willing to work at lightning speeds to avoid delaying the start of a trial.”
This Phase 1 clinical trial, which is currently underway, is the first to study a treatment for disorders caused by mutations in the IGHMBP2 gene, marking another significant milestone in the history of translational research at Nationwide Children’s.
“We have a wonderful multifaceted team here that allows us to transition new experimental therapies to patients as safely and efficiently as possible,” says Megan Waldrop, MD, a pediatric neurologist at Nationwide Children’s and the principal investigator of the current trial.
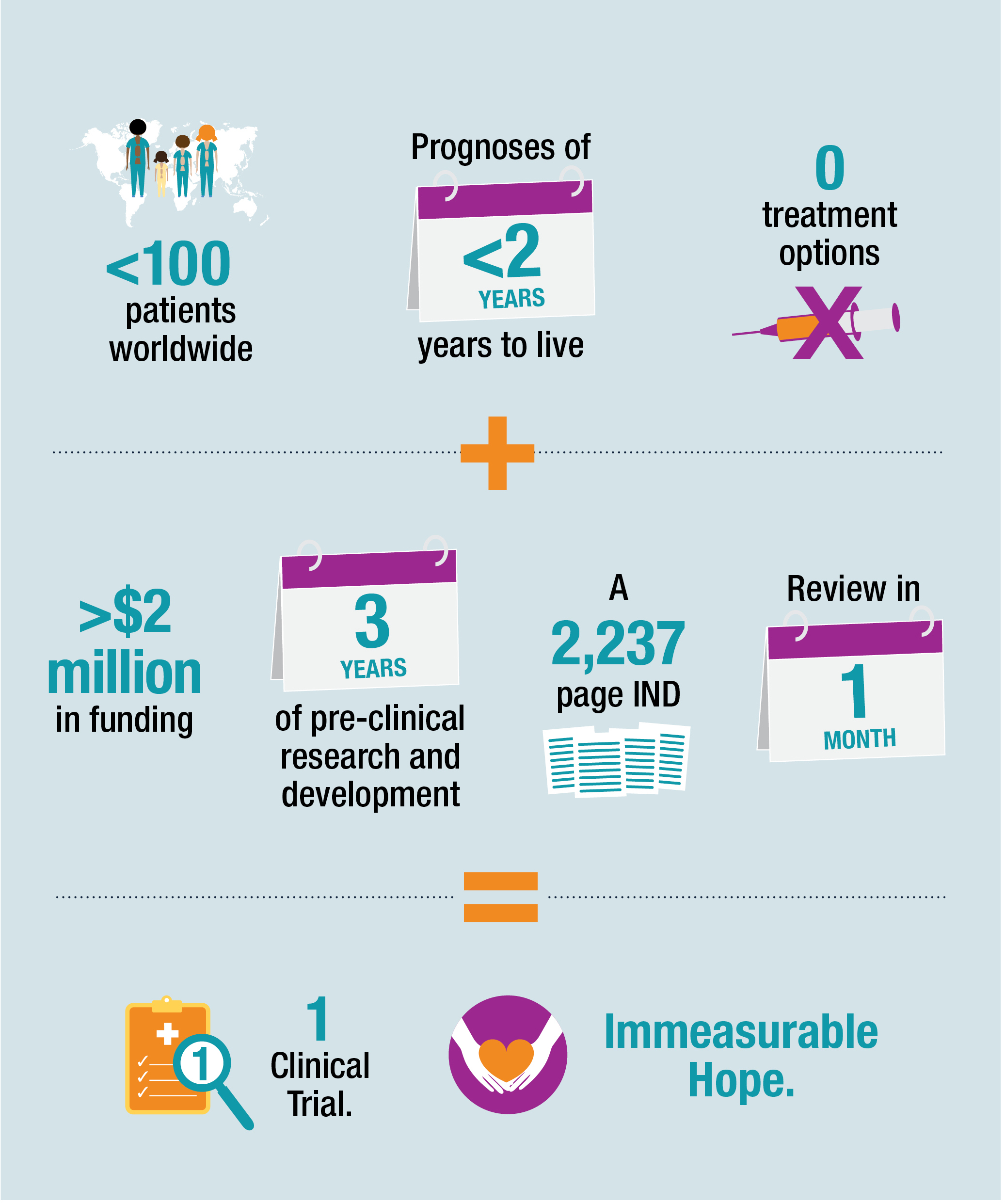
The Difference Innovation Makes
The journey of a new therapeutic — from a discovery or concept to an FDA-approved, commercially-available treatment — is winding and fraught with challenges. The time between when an IND is submitted, when an institution receives notice from the FDA that it may proceed and when actual treatment of patients begins can stretch over months.
“These kinds of things are very complex. There are so many pieces that need to fall in place to keep the program moving forward,” says Dr. Meyer.
Yet for a child with a neurodegenerative disease, waiting weeks to months might mean losing the ability to walk, stand, sit — or even breathe.
In the last seven years, the FDA has greenlit clinical trials for five investigational drugs that grew out of the research programs of Dr. Meyer and Brian Kaspar, PhD, who was a faculty member at Nationwide Children’s for over 13 years, founded gene therapy startups during his tenure with Nationwide Children’s, and made the fundamental discovery that an adeno-associated virus vector could cross the blood brain barrier when injected into the vascular system to deliver genes directly to motor neurons. None of these five trials were delayed by the review process.
“This shows how well-oiled our translational machinery is at Nationwide Children’s,” says Dr. Meyer. “These teams, and their commitment to quality, collaboration and constant improvements — and most of all to patients — are what kept things moving forward at this pace. People here understand there are patients behind the process, and it matters to those patients whether committees meet this week or next week.”
To prepare to submit an IND application for AAV9. IGHMBP2, teams across Nationwide Children’s, including researchers, regulatory staff, sponsored project officers, the Institutional Review Board (IRB) and Institutional Biosafety Committee (IBC), cores and shared resources, licensing team and legal services, as well as numerous external collaborators, were involved in a thorough and constant review process.
“This IND was notably complicated and marks the largest and most thorough regulatory submission Nationwide Children’s has sent to the FDA to date,” says Kevin Bosse, PhD, RAC, who leads the ORRA. “I’m still impressed by the number and quality of preclinical studies performed to support it, the careful and detailed documentation of clinical protocols, and the rigor of this review.”




The Start of the Road
The road to commercial viability for AAV9.IGHMBP2 began in Italy, when Dr. Meyer’s mentor, Dr. Kaspar, and the Corti Lab at Associazione Centro Dino Ferrari headed up a collaborative effort to develop an initial version of a gene therapy vector for treating IGHMBP2-related disease phenotypes — including spinal muscular atrophy with respiratory distress type 1 (SMARD1), a severe disease associated with deterioration of motor neurons of the spinal cord, and Charcot-Marie-Tooth disease type 2S (CMT2S), a milder, but still rare, disease that causes progressive weakness and sensory loss but is less often characterized by significant respiratory compromise early in life.
In 2018, inspired by Dr. Meyer’s track record of translational neurodegenerative disease research and moving therapeutic programs towards clinical trials, a patient foundation, smashSMARD, approached her to pick the project back up.
“When smashSMARD visited to ask me to lead the preclinical development of a gene therapy to treat SMARD1, they told me stories of patients and showed me photos,” says Dr. Meyer. “It was incredibly powerful; I obviously had to help.”
Dr. Meyer and her team examined the initial gene therapy vector that had been developed and modified it to make it more efficient.
Same Gene, Different Disease: SMARD1 vs. CMT2S
As defined by the Orphan Drug Act of 1983, a rare disease is any condition that affects fewer than 200,000 people in the United States. While SMARD1 is the second most common form of spinal muscular atrophy (SMA) and the second most common motor neuron disease of infancy, it affects fewer than 100 patients worldwide. Its uncommonness, however, is of little comfort to patients and families impacted by its traumatizing and devastating pathology.
Within the first six months of life, infants diagnosed with the severe disease may be smaller, move less than expected and begin to show signs of breathing issues. If breathing stops, it can cause or be confused with sudden infant death syndrome (SIDS). Ultimately, infants diagnosed with this severe type of SMA may be permanently paralyzed and unable to survive without a ventilator. Most die before their second birthday if they do not have a feeding tube or breathing tube placed.
“Sadly, when a child gets a SMARD1 diagnosis, families are told by doctors who have never encountered anyone with the disease before, ‘Go home and love your child, because there are literally no options out there,’” says Dr. Meyer. “When no disease-modifying treatment options exist, the impact a single gene therapy could have on these families is profound.”
With an estimated prevalence of one in 2,500, Charcot- Marie-Tooth (CMT) disease is the most common inherited degenerative nerve disease. The milder phenotype caused by IGHMBP2 mutations represents only a small subset of CMT patients. These patients may have typical early development and may not be diagnosed until later in life when gait abnormalities or progressive weakness first appear.
When patients have mutations of the same gene, it can be unclear whether or why one will have milder disease than another. Some children with more severe disease have deletion mutations or mutations that more strongly reduce or even destroy protein function, but the correlation isn’t always perfect. Even patients who are related can present with different clinical phenotypes.
The team, and the FDA, were interested in learning whether a new gene therapy treatment would work across all disease severities. By collaborating with Gregory Cox, PhD, associate professor at The Jackson Laboratory, who cloned the gene for neuromuscular degeneration in a mouse model for SMARD1, and with funding support from smashSMARD and smashSMARD Germany, the teams tested two versions of the vectors in three mouse models with varying disease progression — and they identified a clear lead. The gene therapy worked not only for SMARD1 but also for the milder disease, CMT2S.
All Hands on Deck From Bench to Bedside
Moving towards a trial required a coordinated effort across the Abigail Wexner Research Institute.
The ORRA team, including director Dr. Bosse and regulatory specialist Rachel Manthe-Gross, PhD, oversaw all pre-IND interaction with the FDA. Before the IND was submitted, the ORRA team and Robyn Cunningham, ASQ, CQA, CHRC, director of the Office of Research Compliance and Integrity at Nationwide Children’s, performed an official review of the study reports to ensure all the necessary data and documentation were included in the IND in the correct format.
Early licensing and agreements support during preclinical research was essential. Jocelyn Eidahl, PhD, a senior licensing associate in the Office of Technology Commercialization, and Tabatha Simmons, PhD, director of the Gene Therapy Clinical Research Unit (GT-CRU) at Nationwide Children’s, facilitated discussions and contract negotiations with both smashSMARD and Alcyone Therapeutics Inc., a gene therapy biotech startup that provided funding and non-monetary support of preclinical development including chemistry, manufacturing and control expertise and that is now funding the clinical trial. Dr. Simmons also oversaw budgeting, the manufacture of the clinical vector — the first one produced by Andelyn Biosciences, an affiliate of Nationwide Children’s — and other aspects of clinical trial start up necessary to begin enrollment.
“This process involved essentially every component of the integrated system of bench-to-bedside research and regulatory support that has been developed at Nationwide Children’s over many years,” says Dennis Durbin, MD, MSCE, president of AWRI.
“These teams are incredibly dedicated and passionate about moving research forward into the clinic to help patients,” says Dr. Meyer. “Together, as One Team, we have achieved an incredible milestone to tackle another disease with no other treatment options to date. We can change the lives of many children and families who are suffering.”
Image credits: Nationwide Children’s
About the author
Natalie is a passionate and enthusiastic writer working to highlight the groundbreaking research of the incredible faculty and staff across Nationwide Children's Hospital and the Abigail Wexner Research Institute. Her work at Nationwide Children's marries her past interests and experiences with her passion for helping children thrive and a long-held scientific curiosity that dates back to competing in the Jefferson Lab Science Bowl in middle school. Natalie holds a bachelor’s degree in sociology from Wake Forest University, as well as minors in women's, gender & sexuality studies and interdisciplinary writing. As an undergraduate student, Natalie studied writing and journalism, engaged with anthropological and sociological research with a focus on race and ethnic relations, served as executive editor for the student newspaper, the Old Gold & Black, and gained marketing experience as an intern for a nonprofit entrepreneurial incubator, Winston Starts, as well as by working for Wake Forest University School of Law Office of Communication and Public Relations and its Innocence and Justice Clinic.
- Natalie Wilsonhttps://pediatricsnationwide.org/author/natalie-wilson/

- Natalie Wilsonhttps://pediatricsnationwide.org/author/natalie-wilson/
- Natalie Wilsonhttps://pediatricsnationwide.org/author/natalie-wilson/

- Natalie Wilsonhttps://pediatricsnationwide.org/author/natalie-wilson/